Quick Start¶
Get started with Quantum Pipeline in under 5 minutes by running a simple VQE simulation for the H₂ molecule.
Basic VQE Simulation¶
Step 1: Prepare Molecule Data¶
Create a file h2_molecule.json with the following content:
[
{
"symbols": ["H", "H"],
"coords": [[0.0, 0.0, 0.0], [0.0, 0.0, 0.74]],
"multiplicity": 1,
"charge": 0,
"units": "angstrom",
"masses": [1.008, 1.008]
}
]
Molecule Format
symbols: Atomic symbolscoords: 3D coordinates in specified unitsmultiplicity: Spin multiplicity (1 = singlet, 2 = doublet, etc.)charge: Total molecular chargeunits: Coordinate units (angstromorbohr)masses: Atomic masses (optional)
Step 2: Run VQE Simulation¶
Step 3: View Results¶
The simulation outputs:
2025-06-15 11:33:36,762 - VQERunner - INFO - Processing molecule 1:
Molecule:
Multiplicity: 1
Charge: 0
Unit: Angstrom
Geometry:
H [0.0, 0.0, 0.0]
H [0.0, 0.0, 0.74]
2025-06-15 11:33:36,850 - VQERunner - INFO - Hamiltonian generated in 0.088 seconds.
2025-06-15 11:33:36,873 - VQERunner - INFO - Problem mapped to qubits in 0.023 seconds.
2025-06-15 11:33:36,873 - VQERunner - INFO - Running VQE procedure...
...
2025-06-15 11:33:47,439 - VQESolver - INFO - Simulation completed in 10.47 seconds and 100 iterations.
Final Energy: -1.1372 Hartree
Expected Result
For H₂ with STO-3G basis, the ground state energy should be approximately -1.137 Hartree, which is within chemical accuracy of experimental values.
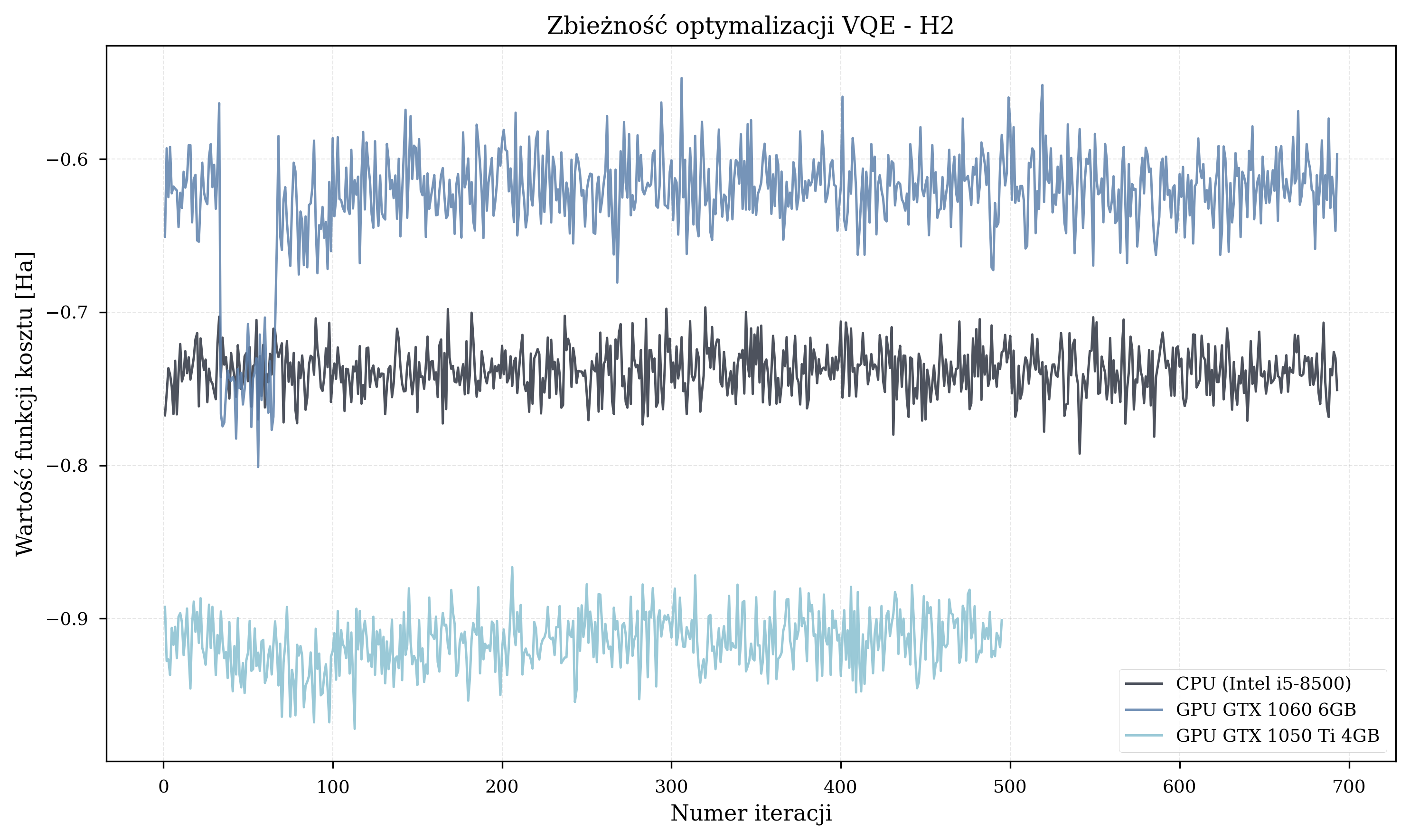
Understanding the Parameters¶
| Parameter | Description | Example |
|---|---|---|
--file |
Path to molecule data file | h2_molecule.json |
--basis |
Basis set for simulation | sto3g, 6-31g, cc-pvdz |
--max-iterations |
Maximum VQE iterations | 100 |
--optimizer |
Optimization algorithm | COBYLA, L-BFGS-B, SLSQP |
--report |
Generate PDF report | Flag (no value) |
See Configuration Guide for all available parameters.
Running Multiple Molecules¶
Create a file with multiple molecules:
[
{
"symbols": ["H", "H"],
"coords": [[0.0, 0.0, 0.0], [0.0, 0.0, 0.74]],
"multiplicity": 1,
"charge": 0,
"units": "angstrom"
},
{
"symbols": ["O", "H", "H"],
"coords": [[0.0, 0.0, 0.0], [0.0, 0.757, 0.586], [0.0, -0.757, 0.586]],
"multiplicity": 1,
"charge": 0,
"units": "angstrom"
}
]
Run the simulation:
python quantum_pipeline.py \
--file molecules.json \
--basis sto3g \
--max-iterations 150 \
--optimizer L-BFGS-B
Convergence-Based Optimization¶
Instead of fixed iterations, use convergence threshold:
python quantum_pipeline.py \
--file h2_molecule.json \
--basis sto3g \
--convergence --threshold 1e-6 \
--optimizer L-BFGS-B
Mutually Exclusive
Do not use both --max-iterations and --convergence together.
Enabling GPU Acceleration¶
If you have an NVIDIA GPU:
python quantum_pipeline.py \
--file h2_molecule.json \
--basis sto3g \
--max-iterations 100 \
--gpu \
--simulation-method statevector
Or with Docker:
docker run --rm --gpus all \
straightchlorine/quantum-pipeline:latest-gpu \
--file /app/data/h2_molecule.json \
--gpu \
--max-iterations 100
See GPU Acceleration Guide for setup.
Streaming Results to Kafka¶
Enable real-time data streaming:
Kafka Required
This requires Kafka and Schema Registry to be running. See Kafka Streaming Guide.
Generating Reports¶
Generate a PDF report with visualizations:
python quantum_pipeline.py \
--file h2_molecule.json \
--basis sto3g \
--max-iterations 100 \
--report
The report includes:
- Molecular structure visualization
- Energy convergence plot
- Hamiltonian coefficients
- Optimal parameters
- Timing breakdown
Example: Advanced Configuration¶
A more advanced example with custom parameters:
python quantum_pipeline.py \
--file molecules.json \
--basis cc-pvdz \
--max-iterations 200 \
--optimizer L-BFGS-B \
--ansatz-reps 5 \
--shots 2048 \
--optimization-level 3 \
--simulation-method statevector \
--report \
--kafka \
--log-level DEBUG
This configuration:
- Uses a larger basis set (
cc-pvdz) for higher accuracy - Allows more iterations for convergence
- Uses gradient-based optimizer (
L-BFGS-B) - Increases ansatz depth (
ansatz-reps=5) - Enables both reporting and Kafka streaming
- Sets verbose logging
What's Next?¶
Now that you've run your first simulation:
- Explore Configuration Options - Learn about all parameters
- Choose the Right Optimizer - 16 optimizers available
- Understand System Architecture - How it all works
- Deploy Full Platform - Run with data engineering
- Monitor Performance - Track system metrics
Common Issues¶
Simulation takes too long
- Try using fixed iterations instead of
--convergence - Choose a faster optimizer like
L-BFGS-B - Start with smaller basis sets like
sto3g
Memory errors with large molecules
- Use
--simulation-method matrix_product_statefor memory efficiency - Reduce
--ansatz-repsto decrease circuit depth - Consider using a smaller basis set
- Enable GPU to offload memory to VRAM (if available)
Results don't match reference energies
- This can also be ansatz initialization issue - currently it's random, which may cause process to get stuck in local minima. This issue is something to be addressed in the future.
- Increase
--max-iterations(try 200-500) - Use tighter convergence threshold:
--convergence --threshold 1e-8 - Try different optimizers (L-BFGS-B recommended)
- Use larger basis sets for better accuracy
See Troubleshooting Guide for more help.