Basis Sets¶
A basis set defines the mathematical functions used to approximate molecular orbitals in quantum chemistry calculations. For VQE simulations, the basis set governs the number of qubits required under the Jordan-Wigner transformation, establishing a trade-off between accuracy and computational cost. This page describes the three basis sets supported by the Quantum Pipeline - STO-3G, 6-31G, and cc-pVDZ - and provides guidance on their selection.
STO-3G¶
Slater-Type Orbital - 3 Gaussian (STO-3G), introduced by Hehre, Stewart, and Pople (1969), is the simplest and most widely used minimal basis set. It approximates each Slater-type orbital (STO) with a linear combination of three Gaussian-type functions (GTOs).
Characteristics¶
- Type: Minimal basis set
- Construction: Each atomic orbital is represented by exactly one basis function, which is itself a contraction of three Gaussian primitives.
- Accuracy: Provides qualitatively correct results for molecular geometries and relative energies, but exhibits significant quantitative errors. Typical energy errors range from 0.1 to 1.0 Ha relative to the complete basis set limit.
- Computational cost: Lowest among all supported basis sets. The small number of basis functions translates directly to fewer qubits and shorter circuit depths.
When to Use STO-3G¶
- Rapid prototyping and algorithm testing
- Benchmarking optimizer performance across many configurations
- Simulations on real quantum hardware where qubit count is severely constrained
- Initial exploration of large molecular systems where higher-accuracy basis sets are computationally prohibitive
6-31G¶
The 6-31G basis set, developed by Ditchfield, Hehre, and Pople (1971), is a split-valence basis set that provides a meaningful improvement in accuracy over STO-3G while maintaining moderate computational cost.
Characteristics¶
- Type: Split-valence double-zeta
- Construction: Core orbitals are represented by a single basis function contracted from six Gaussian primitives. Valence orbitals are split into two components: an inner part contracted from three Gaussians and an outer part consisting of a single Gaussian. This splitting allows the valence electron density to adjust its spatial extent during the self-consistent field procedure.
- Accuracy: Substantially better than STO-3G for energies, geometries, and other molecular properties. Typical energy errors range from 0.01 to 0.1 Ha.
- Computational cost: Moderate - estimated at approximately 2 to 5 times slower than STO-3G based on basis set literature (no 6-31G timing experiments were conducted in the pipeline).
When to Use 6-31G¶
- Standard calculations on small-to-medium molecules where STO-3G accuracy is insufficient
- Studies requiring a reasonable balance between accuracy and computational feasibility
- Investigations of basis set convergence trends without the full expense of correlation-consistent sets
CC-pVDZ¶
The correlation-consistent polarized Valence Double-Zeta (cc-pVDZ) basis set, introduced by Dunning (1989), represents a significant step up in accuracy and computational demands. It is designed specifically for correlated calculations and forms part of a systematic hierarchy (cc-pVDZ, cc-pVTZ, cc-pVQZ, ...) that converges smoothly toward the complete basis set limit.
Characteristics¶
- Type: Correlation-consistent polarized double-zeta
- Construction: Includes polarization functions (higher angular momentum functions beyond those occupied in the atomic ground state) that are essential for capturing electron correlation effects. The basis functions are optimized to recover correlation energy systematically.
- Accuracy: Significantly superior to both STO-3G and 6-31G. Typical energy errors range from 0.001 to 0.01 Ha. Well-suited for quantitative comparison with experimental results and high-level classical methods.
- Computational cost: High - approximately 10 to 100 times slower than STO-3G. The increased number of basis functions leads to substantially more qubits under the Jordan-Wigner mapping.
When to Use CC-pVDZ¶
- High-accuracy quantum chemistry calculations requiring quantitative agreement with experiment
- Studies of electron correlation effects
- Validation of VQE results against established classical methods (CCSD, FCI)
- Research applications where computational cost is secondary to accuracy
Comparison¶
The following table summarizes the key properties of each supported basis set, including qubit requirements for representative molecular systems as determined by experiments conducted with the Quantum Pipeline framework.
| Property | STO-3G | 6-31G | cc-pVDZ |
|---|---|---|---|
| Type | Minimal | Split-valence | Correlation-consistent |
| Accuracy | Basic (\(\sim\)0.1-1 Ha error) | Moderate (\(\sim\)0.01-0.1 Ha error) | High (\(\sim\)0.001-0.01 Ha error) |
| Qubits (H\(_2\)) | 4 | 8 | 10 |
| Qubits (H\(_2\)O) | 14 | 26 | 28 |
| Relative Cost | 1x (baseline) | 2-5x | 10-100x |
| Memory Scaling | Minimal | Moderate | High (often >10x STO-3G) |
| Polarization Functions | No | No | Yes |
| Correlation Recovery | Poor | Partial | Systematic |
Qubit counts under Jordan-Wigner mapping
The qubit counts listed above correspond to the number of spin-orbitals produced by each basis set under the Jordan-Wigner transformation. The STO-3G values are empirically verified through the pipeline's PySCF/Jordan-Wigner integration (used in all primary experiments). The 6-31G and cc-pVDZ values are consistent with standard Jordan-Wigner mapping for these basis sets (Szabo & Ostlund 1996). Exact counts may vary depending on symmetry reduction, active space selection, and frozen-core approximations applied during Hamiltonian construction.
Accuracy range sources
The typical energy error ranges reported above are established in the original basis set literature: STO-3G ~0.1-1 Ha (Hehre et al. 1969; Szabo & Ostlund 1996), 6-31G ~0.01-0.1 Ha (Ditchfield, Hehre & Pople 1971), and cc-pVDZ ~0.001-0.01 Ha (Dunning 1989). These ranges represent errors relative to the complete basis set limit and vary with the molecular system and level of electron correlation treatment.
Selection Guide¶
The appropriate basis set depends on the objectives and constraints of the simulation. The following recommendations are drawn from experimental experience with the Quantum Pipeline framework.
| Use Case | Recommended Basis Set | Rationale |
|---|---|---|
| Rapid prototyping and testing | STO-3G | Fastest execution, minimal resource requirements |
| Standard small-molecule calculations | 6-31G | Good accuracy-cost balance |
| High-accuracy quantum chemistry | cc-pVDZ | Systematic correlation recovery |
| VQE algorithm benchmarking | STO-3G | Fast, repeatable results across many configurations |
| Comparison with experimental data | cc-pVDZ or higher | Quantitative accuracy required |
| Real quantum hardware experiments | STO-3G | Minimizes qubit count and circuit depth |
| Large molecules (>10 atoms) | STO-3G or 6-31G | Computational feasibility constraints |
Decision Flowchart¶
When selecting a basis set, consider the following progression:
- Start with STO-3G to verify that the simulation pipeline functions correctly and to establish baseline results.
- Upgrade to 6-31G if quantitative improvements are needed and computational resources permit.
- Use cc-pVDZ only when high accuracy is essential and the molecular system is small enough to remain tractable.
Impact on VQE Performance¶
Basis set choice affects VQE performance. The following data is from thesis experiments with the pipeline.
Optimization Results¶
cc-pVDZ experiments on H₂ showed harder convergence than STO-3G:
- With STO-3G (4 qubits), the H\(_2\) optimizer terminated after approximately 650 iterations on average, though the best result (-1.07 Ha) was still 4.2% above the reference value (-1.117 Ha).
- With cc-pVDZ (10 qubits), the same molecule failed to converge to the correct ground-state energy within the allocated iterations, yielding energies of approximately 24-26 Ha versus the expected value of approximately -1 Ha.
This is due to the larger parameter space under random initialization.
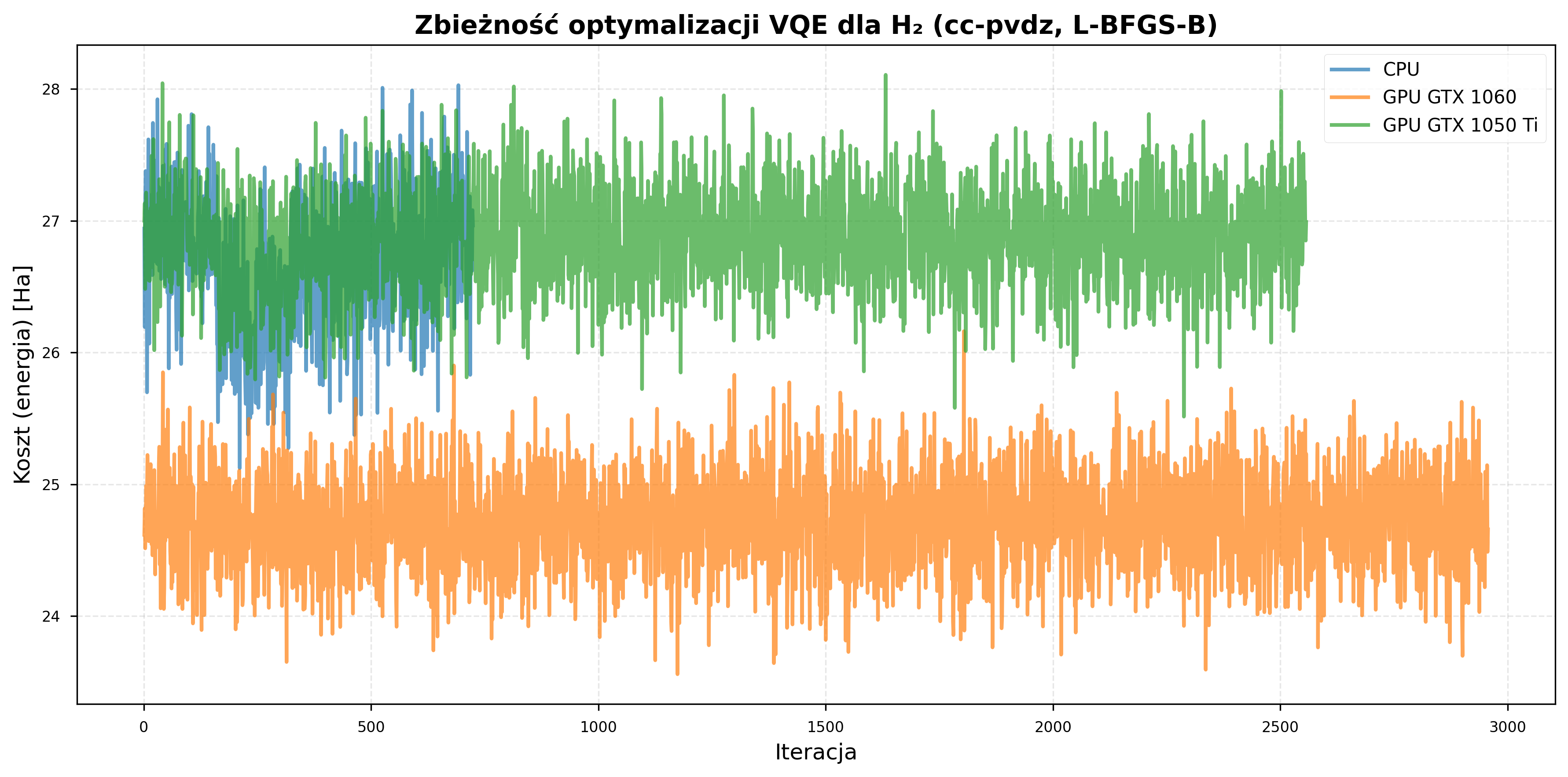
Iteration Timing¶
The computational cost per iteration scales with basis set size. Measured iteration times for H\(_2\):
| Configuration | STO-3G (s/iter) | cc-pVDZ (s/iter) | Slowdown Factor |
|---|---|---|---|
| CPU (i5-8500) | ~4.3 | 206.2 | ~48x |
| GPU GTX 1060 | ~2.4 | 50.6 | ~21x |
| GPU GTX 1050 Ti | ~2.5 | 58.4 | ~23x |
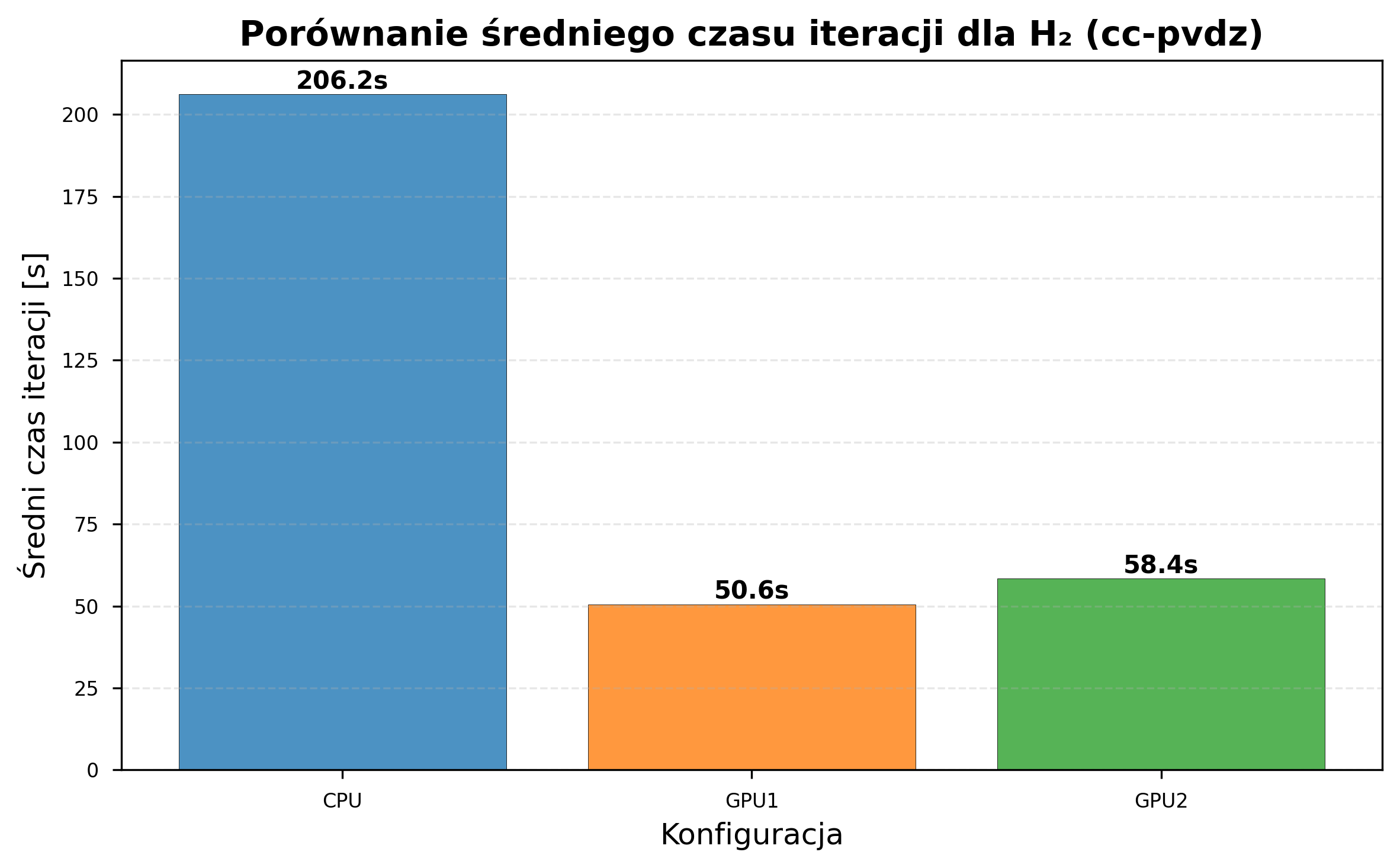
GPU Acceleration Scaling¶
GPU acceleration benefits increase with basis set complexity: STO-3G sees 1.74-1.81x speedup over CPU, while cc-pVDZ sees 3.53-4.08x due to greater parallelizable matrix operations.
Based on the cc-pVDZ timing data (206s/iter on CPU vs. 50s/iter on GPU), GPU acceleration appears necessary for practical execution times with larger basis sets. However, cc-pVDZ was only tested on H\(_2\); larger molecules were not benchmarked with this basis set.
Implementation Details¶
Basis sets in the Quantum Pipeline are managed through integration with the PySCF library. The supported basis sets are defined in the system configuration:
The basis set is passed to the PySCF driver during Hamiltonian construction:
driver = PySCFDriver.from_molecule(molecule, basis=self.basis_set)
problem = driver.run()
hamiltonian = problem.second_q_ops()[0]
Additional basis sets supported by PySCF (e.g., cc-pVTZ, aug-cc-pVDZ,
6-311++G**) can be added by extending the SUPPORTED_BASIS_SETS list without
modifying the core simulation logic.
Summary¶
The pipeline supports three basis sets with different accuracy-cost trade-offs. STO-3G provides a computationally efficient baseline suitable for algorithm development and rapid prototyping. 6-31G offers a meaningful accuracy improvement at moderate additional cost. cc-pVDZ delivers correlation-consistent accuracy but demands substantially greater computational resources, making GPU acceleration particularly valuable. The Quantum Pipeline framework supports all three basis sets and is extensible to additional sets through its PySCF integration.
References¶
- Szabo, A. & Ostlund, N.S. Modern Quantum Chemistry: Introduction to Advanced Electronic Structure Theory. Dover Publications (1996)
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 90, 1007-1023 (1989)
- Hehre, W.J. et al. Self-Consistent Molecular-Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 56, 2257-2261 (1972)
- PySCF Documentation - Basis Sets