Basic Usage¶
CLI syntax¶
Only --file is required. All other flags have defaults defined in
defaults.py.
To process a single molecule by its 0-based index:
Common workflows¶
Configuration files¶
--dump saves the current CLI configuration. It writes a JSON file to
run_configs/ with a name built from the molecule file, basis set, optimizer,
backend, and date (for example molecules-cc-pvdz-L-BFGS-B-local-20260613.json):
quantum-pipeline \
--file data/molecules.json \
--basis cc-pvdz \
--max-iterations 200 \
--optimizer L-BFGS-B \
--dump
--load points the run at a saved configuration file and validates it:
The loaded values are not currently applied, though: the run still uses the
arguments and defaults from the command line, so treat --dump as a record of a
run rather than a way to replay one. --dump and --load cannot be used together.
The dumped JSON mirrors the CLI arguments. For the field names, see
defaults.py.
Ansatz and initialization¶
Three ansatz types are supported: EfficientSU2 (default), RealAmplitudes, and ExcitationPreserving.
Two initialization strategies are available:
| Strategy | Flag | Description |
|---|---|---|
| Random | --init-strategy random |
Uniform [0, 2pi]. Default, but can trap in local minima. |
| Hartree-Fock | --init-strategy hf |
Uses HF parameters as a starting point. More reliable convergence, especially with cc-pvdz. Only works with EfficientSU2. |
Simulation methods¶
The --simulation-method flag selects the Aer simulator backend:
| Method | Description |
|---|---|
statevector |
Dense statevector simulation (default) |
automatic |
Aer selects the best method based on circuit and noise model |
density_matrix |
Dense density matrix, for noisy circuits |
stabilizer |
Clifford stabilizer simulator |
extended_stabilizer |
Approximate Clifford+T simulator |
matrix_product_state |
Tensor-network MPS, lower memory for large circuits |
unitary |
Computes the unitary matrix (no measurement) |
superop |
Dense superoperator matrix |
tensor_network |
GPU-only, requires cuTensorNet |
tensor_network requires the --gpu flag.
Output structure¶
Results are saved under the gen/ directory. The --output-dir flag is accepted but currently has no effect; output always goes to gen/.
gen/
graphs/
molecule_plots/
operator_plots/
complex_operator_plots/
energy_plots/
ansatz/
ansatz_decomposed/
performance_metrics/
*.pdf
PDF reports are generated when --report is passed. See an
example report.
The ansatz/ and ansatz_decomposed/ directories contain circuit diagrams that
often are too large to fit into the PDF, especially for larger molecules.
See ansatz and
decomposed ansatz
for H2.
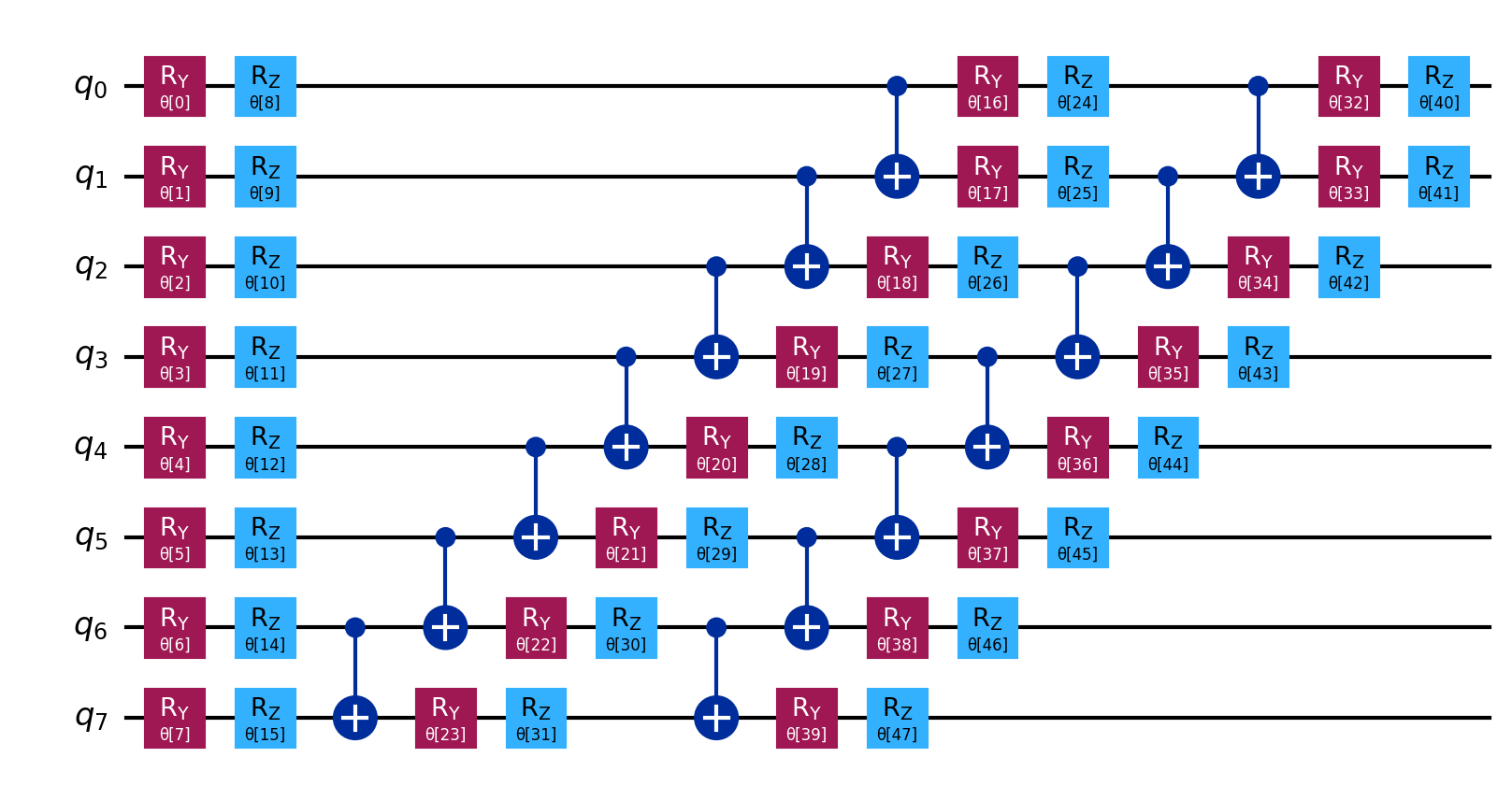{kind=link}
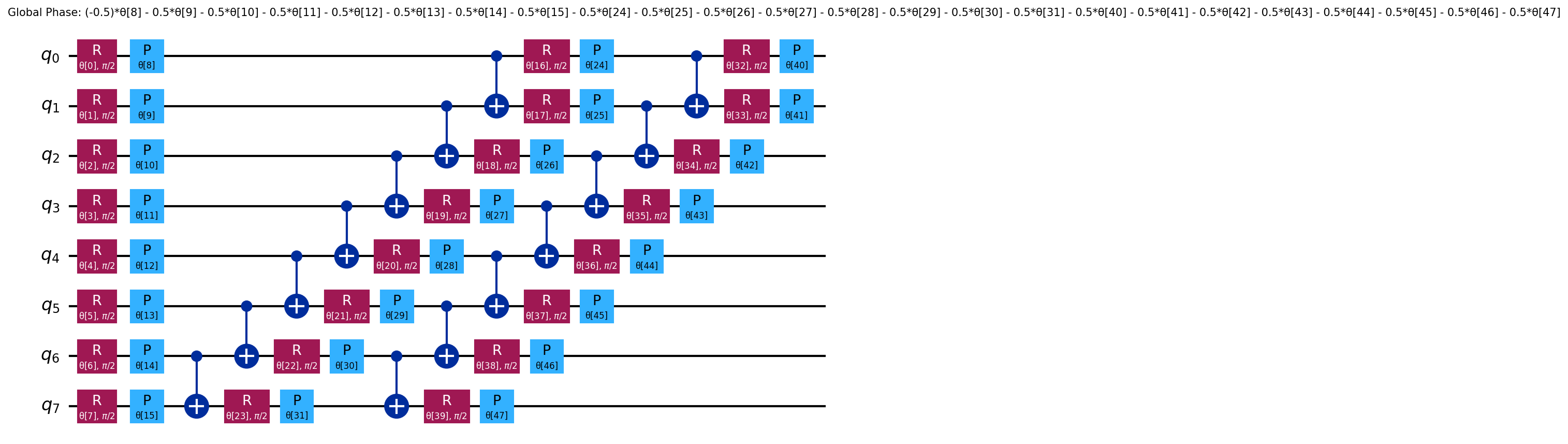{kind=link}
Log levels¶
Control verbosity with --log-level:
# Minimal output
quantum-pipeline --file data/molecules.json --log-level ERROR
# Default
quantum-pipeline --file data/molecules.json --log-level INFO
# Verbose
quantum-pipeline --file data/molecules.json --log-level DEBUG
Performance monitoring¶
Enable system resource tracking during simulation:
quantum-pipeline \
--file data/molecules.json \
--enable-performance-monitoring \
--performance-interval 30 \
--performance-pushgateway http://localhost:9091 \
--performance-export-format both
Or via environment variables:
export MONITORING_ENABLED=true
export PUSHGATEWAY_URL=http://localhost:9091
export MONITORING_INTERVAL=10
export MONITORING_EXPORT_FORMAT=both
quantum-pipeline --file data/molecules.json
See Monitoring for more.
Recipes - basic parameter combinations¶
Fast prototyping
quantum-pipeline \
--file data/molecules.json \
--basis sto3g \
--max-iterations 50 \
--optimizer COBYLA
Balanced
quantum-pipeline \
--file data/molecules.json \
--basis sto3g \
--max-iterations 150 \
--optimizer L-BFGS-B \
--ansatz-reps 2
High accuracy
quantum-pipeline \
--file data/molecules.json \
--basis cc-pvdz \
--convergence \
--threshold 1e-8 \
--optimizer L-BFGS-B \
--init-strategy hf \
--ansatz-reps 5
Python API¶
The pipeline can also be used programmatically. The VQERunner
class accepts parameters similar to the CLI:
from quantum_pipeline.runners.vqe_runner import VQERunner
runner = VQERunner(
filepath='data/molecules.json',
basis_set='sto3g',
max_iterations=100,
optimizer='L-BFGS-B',
init_strategy='hf',
seed=42,
)
runner.run()
# Results are stored per molecule
for result in runner.run_results:
print(f'{result.basis_set}: {result.vqe_result.minimum:.6f} Ha')
print(f' Iterations: {len(result.vqe_result.iteration_list)}')
print(f' Total time: {result.total_time:.2f}s')
The constructor defaults differ from the CLI: when you omit them it uses the
COBYLA optimizer (CLI default is L-BFGS-B) and ansatz_reps=3 (CLI default is
2). Pass these explicitly to reproduce a CLI run. For the full constructor
signature, see the source.
Next steps¶
- Configuration Reference - all available parameters
- Optimizer Guide - choosing the right optimizer
- Simulation Methods - backend details
- Examples - more use cases