Variational Quantum Eigensolver¶
The Variational Quantum Eigensolver (VQE) is a hybrid quantum-classical algorithm designed to approximate the ground-state energy of a quantum system. Originally proposed by Peruzzo et al. (2014), VQE combines parameterized quantum circuits with classical optimization to find the lowest eigenvalue of a molecular Hamiltonian. Its shallow circuit depth and tolerance for noise make it one of the most practical algorithms for near-term quantum devices operating in the NISQ (Noisy Intermediate-Scale Quantum) regime.
For a thorough treatment of VQE theory, see Tilly et al. (2022) and the Qiskit VQE tutorial.
Algorithm Overview¶
The VQE algorithm operates as an iterative loop between a quantum processor (or simulator) and a classical optimizer:
-
Initialization - Select a molecular system, basis set, and ansatz. Initialize the variational parameters \(\theta\).
-
State Preparation - Execute the parameterized quantum circuit (ansatz) to prepare the trial state \(\lvert \psi(\theta) \rangle\).
-
Energy Measurement - Measure the expectation value \(\langle \psi(\theta) | \hat{H} | \psi(\theta) \rangle\) by decomposing the Hamiltonian into a sum of Pauli operators.
-
Classical Optimization - Feed the measured energy back to a classical optimizer (e.g., L-BFGS-B, COBYLA), which proposes updated parameters \(\theta'\).
-
Convergence Check - If the energy change between successive iterations falls below a specified threshold (e.g., \(10^{-6}\) Ha), terminate. Otherwise, return to step 2.
Flowchart¶
The following diagram illustrates the VQE optimization loop as implemented in the Quantum Pipeline:
flowchart TD
A["Initialize parameters #952;"] --> B["Prepare state #124;#968;#9002; via ansatz"]
B --> C["Measure #9001;#968;#124;H#124;#968;#9002;"]
C --> D[Return energy to classical optimizer]
D --> E{Converged?}
E -->|No| F["Update #952; #8594; #952;#39;"]
F --> B
E -->|Yes| G["Report ground-state energy E#8320;"]
style A fill:#7986cb,color:#ffffff
style G fill:#66bb6a,color:#ffffff
style E fill:#ffb74d,color:#ffffffAnsatz Construction¶
The ansatz is the parameterized quantum circuit that prepares the trial state. The choice of ansatz is critical to VQE performance: it must be expressive enough to represent the ground state while remaining shallow enough to execute on noisy hardware.
UCCSD Ansatz¶
The Unitary Coupled Cluster Singles and Doubles (UCCSD) ansatz is a chemistry-inspired construction that applies single and double excitation operators to a Hartree-Fock reference state:
While UCCSD provides strong theoretical guarantees, its circuit depth can be prohibitive for NISQ devices, motivating hardware-efficient alternatives.
EfficientSU2 Ansatz¶
The Quantum Pipeline employs the EfficientSU2 ansatz from Qiskit (source) as the default circuit construction. EfficientSU2 is a hardware-efficient ansatz that uses layers of single-qubit SU(2) rotations followed by entangling CNOT gates. Its advantages include:
- Shallow circuit depth - scales linearly with the number of qubits and layers, making it feasible for NISQ simulation.
- Full SU(2) coverage - each qubit undergoes RY and RZ rotations, providing sufficient expressibility for many molecular systems.
- Flexible entanglement - supports various entanglement patterns (linear, full, circular).
The trade-off is that hardware-efficient ansatze lack the physical intuition of UCCSD and may encounter optimization difficulties such as barren plateaus for large systems.
Experimental Observations¶
The thesis experiments ran VQE with random parameter initialization and a single optimizer (L-BFGS-B) across six molecules. The results illustrate both the potential and the current limitations of the approach.
The optimizer ran for approximately 650 iterations (H\(_2\)) and 630 iterations (HeH\(^+\)) on average for 4-qubit systems, and 1,500-2,700 iterations for larger molecules (8-12 qubits). In most cases, the optimizer was terminated without reaching the known ground-state energy — the runs show the optimizer exploring the landscape and getting trapped in local minima, not converging to the correct solution.
Why the Results Fall Short¶
The pipeline initializes EfficientSU2 parameters from a uniform random
distribution over \([0, 2\pi)\) via
np.random.random().
Because the VQE cost function is non-convex and L-BFGS-B is a local optimizer,
each run converges to the nearest minimum from its starting point - not
necessarily the global minimum.
- Small molecules (H\(_2\), 4 qubits, 32 parameters): Figure 1 shows one of three runs approaching -1.117 Ha (HF/STO-3G; Szabo & Ostlund 1996, p.108) while the other two settle in shallower local minima.
- Larger molecules (H\(_2\)O, 12 qubits, 96+ parameters): Random starting points produced relative errors of 9-25% in the benchmarking results.
EfficientSU2 does not preserve particle number or spin symmetry, which can lead to anomalous results such as the HeH\(^+\) VQE energy falling below the exact Full CI value (see Benchmarking: Comparison with Reference Values).
Hardware-efficient ansatze are also susceptible to barren plateaus - regions where gradients vanish exponentially with system size (McClean et al. 2018).
The following are planned next steps to address the problems documented above:
- Hartree-Fock-informed initialization - using the classical HF solution as a starting point instead of random parameters.
- Adaptive ansatze (ADAPT-VQE) - dynamically growing the circuit to lower energy at each step (Grimsley et al. 2019).
- Multiple random restarts - running VQE from several initial points and selecting the best result.
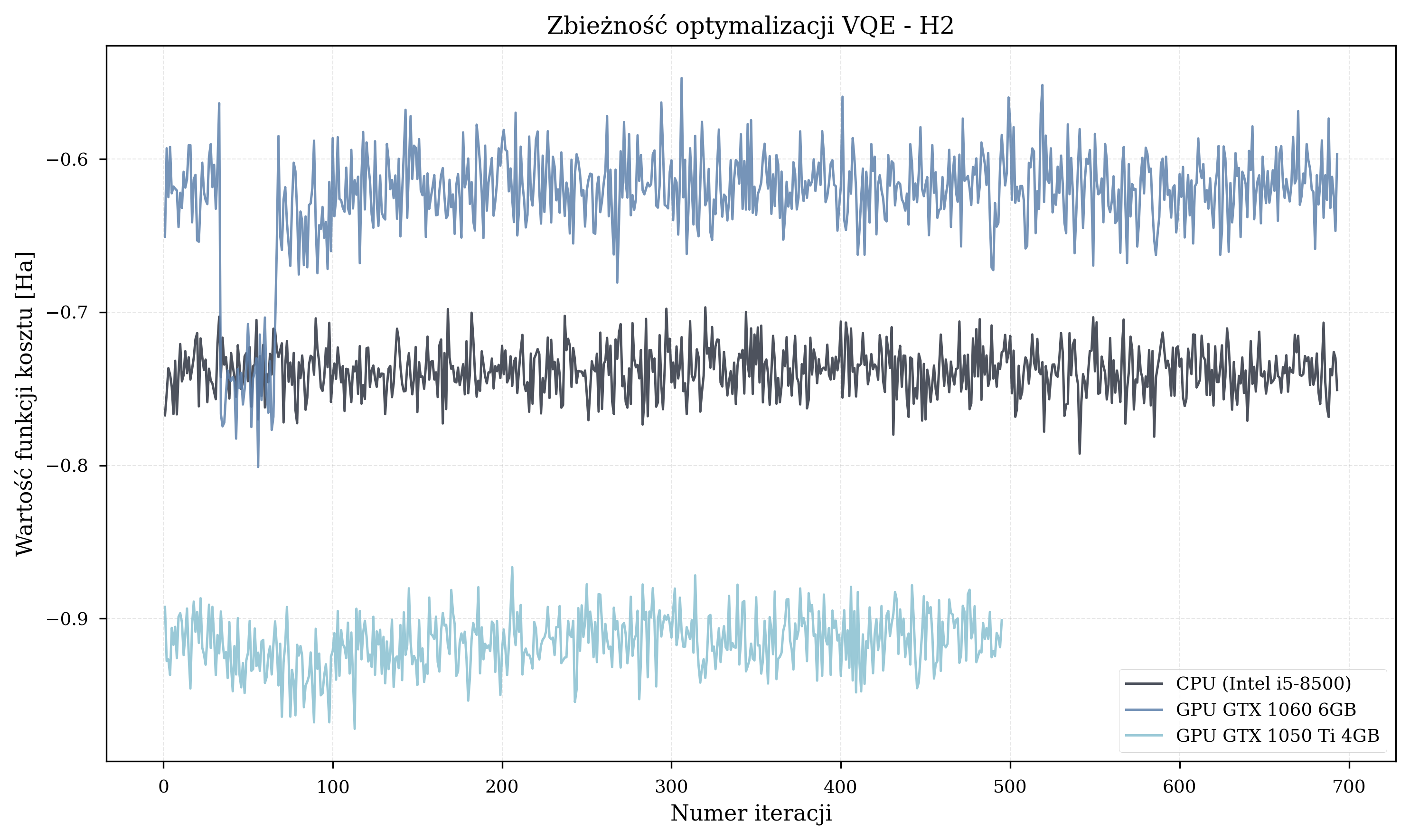
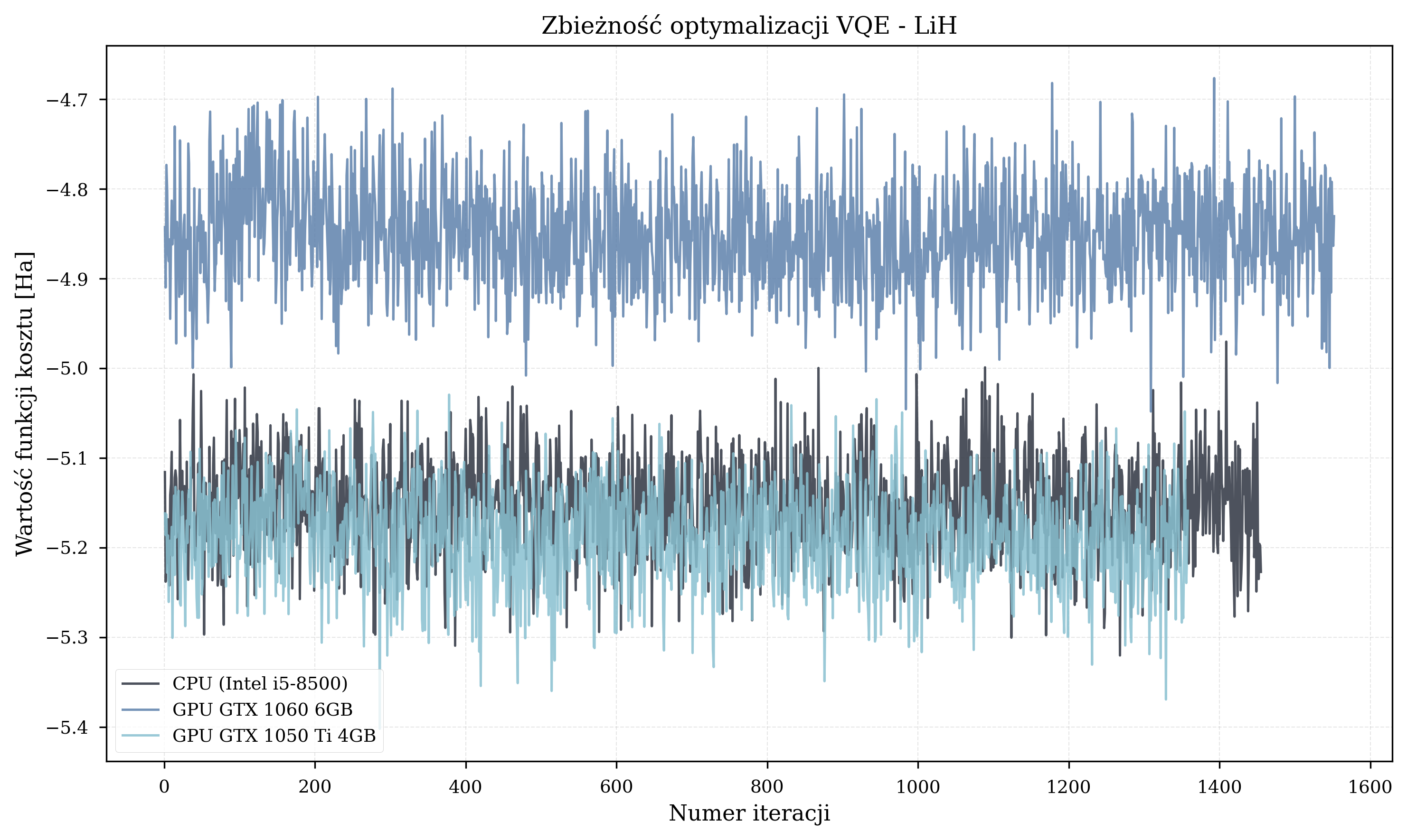
Known problems identified in the thesis experiments:
- Basis set complexity — cc-pVDZ experiments failed entirely (energies of 24-26 Ha vs expected -1 Ha for H\(_2\)).
- Ansatz limitations — EfficientSU2 does not preserve particle number or spin symmetry, leading to unphysical results (e.g. HeH\(^+\) sub-FCI anomaly).
Implementation in Quantum Pipeline¶
Within the Quantum Pipeline framework, VQE simulations are executed through the
vqe_runner module, which orchestrates the interaction between Qiskit's quantum
circuit primitives and the classical optimization backend. Key implementation
details include:
- Hamiltonian construction via PySCF driver integration, supporting multiple basis sets and molecular geometries.
- Qubit mapping via Jordan-Wigner transformation, converting the second-quantized Hamiltonian to a qubit operator.
- Ansatz selection defaulting to EfficientSU2 (source) with configurable depth and entanglement topology.
- Optimizer defaulting to L-BFGS-B
(source),
with support for COBYLA,
SLSQP,
Nelder-Mead,
and SPSA. All optimizers are provided by
scipy.optimize.minimize. See the Optimizers page for configuration details. - Statevector simulation with optional GPU acceleration through NVIDIA cuQuantum, enabling significant speedups for medium-to-large molecular systems (see GPU Acceleration).
- Streaming telemetry - iteration-level data (energy, parameters, timing) is published to Apache Kafka for real-time monitoring and post-hoc analysis.
For practical guidance on running VQE simulations, consult the Quick Start and Examples pages.
References¶
- Peruzzo, A. et al. A variational eigenvalue solver on a photonic quantum processor. Nature Communications 5, 4213 (2014).
- McClean, J.R. et al. The theory of variational hybrid quantum-classical algorithms. New Journal of Physics 18, 023023 (2016).
- Tilly, J. et al. The Variational Quantum Eigensolver: A review of methods and best practices. Physics Reports 986, 1-128 (2022).
- McClean, J.R. et al. Barren plateaus in quantum neural network training landscapes. Nature Communications 9, 4812 (2018).
- Grimsley, H.R. et al. An adaptive variational algorithm for exact molecular simulations on a quantum computer. Nature Communications 10, 3007 (2019).
- Szabo, A. & Ostlund, N.S. Modern Quantum Chemistry: Introduction to Advanced Electronic Structure Theory. Dover Publications (1996).